New tentative approval for Xiamen Lp drug topiramate
[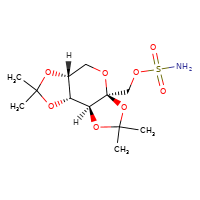](//www.DrugPatentWatch.com/p/preview/generic-api/topiramate?utm_medium=dpw_wp_blog&utm_campaign=dpw_wp_blog&utm_source=dpw_wp_blog) Topiramate is the generic in…
